Лиганды - ионы или молекулы, которые непосредственно связаны с комплексообразователем и являются донорами электронных пар. Эти электроноизбыточные системы, имеющие свободные и подвижные электронные пары, могут быть донорами электронов, например: Соединения р-элементов проявляют комплексообразующие свойства и выступают в комплексном соединении в качестве лигандов. Лигандами могут быть атомы и молекулы
(белка, аминокислот, нуклеиновых кислот, углеводов). Эффективность и прочность донорно-акцкпторного взаимодействия лиганда и комплексообразователя определяется их поляризуемостью-способностью частицы трансформировать свои электронные оболочки под внешним воздействием.
Константа нестойкости:
Кнест= 2 /
К уст=1/Кнест
Реакции замещения лигандов
Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Yс комплексом – происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев – замещение лиганда LрастворителемS, который далее легко замещается молекулой субстратаY
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
Представления о строении металлоферментов и других биокомплексных соединений (гемоглобин, цитохромы, кобаламины). Физико-химические принципы транспорта кислорода гемоглобином.
Особенности строения металлоферментов.
Биокомплексные соединения значительно различаются по устойчивости. Роль металла в таких комплексах высокоспецифична: замена его даже на близкий по свойствам элемент приводит к значительной или полной утрате физиологической активности.
1. В12: содержит 4 пиррольных кольца,ион кобальта и группы CN-. Способствует переносу атома H на атом С в обмен на какую либо группу, участвует в процессе образования дезоксирибозы из рибозы.
2. гемоглобин:имеет четвертичную структуру. Четыре полипептидные цепи, соединённые вместе, образуют почти правильную форму шара, где каждая цепь контактирует с двумя цепями.
Гемоглобин
- дыхательный пигмент, придающий крови красный цвет. Гемоглобин состоит из белка и железопорфирина и переносит кислород от органов дыхания к тканям тела и углекислый газ от них к дыхательным органам.
Цитохромы
- сложные белки (гемопротеиды), осуществляющие в живых клетках ступенчатый перенос электронов и/или водорода от окисляемых органических веществ к молекулярному кислороду. При этом образуется богатое энергией соединение АТФ.
Кобаламины
- природные биологически активные кобальторганические соединения. Структурной основой К. является корриновое кольцо, состоящее из 4 пиррольных ядер, у которых атомы азота связаны с центральным атомом кобальта.
Физико-химические принципы транспорта кислорода гемоглобином - Атом (Fe (II)) (один из компонентов гемоглобина) способен образовывать 6 координационных связей. Из них четыре используются для закрепления самого атома Fe(II) в геме, пятая связь - для связывания гема с белковой субъединицей, а с помощью шестой связи происходит связывание молекулы О 2 или СО 2.
Металло-лигандный гомеостаз и причины его нарушения. Механизм токсического действия тяжелых металлов и мышьяка на основе теории жестких и мягких кислот и оснований (ЖМКО). Термодинамические принципы хелатотерапии. Механизм цитотоксического действия соединений платины.
В организме непрерывно происходит образование и разрушение биокомплексов из катионов металлов и биолигандов (порфинов, аминокислот, белков, полинуклеотидов), в состав которых входят донорные атомы кислорода, азота, серы. Обмен с окружающей средой поддерживает концентрации этих веществ на постоянном уровне, обеспечивая металло-лигандный гомеостаз . Нарушение сложившегося равновесия ведет к ряду патологических явлений – металлоизбыточным и металлодефицитным состояниям. В качестве примера можно привести неполный перечень заболеваний, связанных с изменением металло-лигандного баланса только для одного иона – катиона меди. Дефицит этого элемента в организме вызывает синдром Менкеса, синдром Морфана, болезнь Вильсона-Коновалова, цирроз печени, эмфизему лёгких, аорто- и артериопатии, анемии. Избыточное поступление катиона может вести к серии заболеваний самых разных органов: ревматизму, бронхиальной астме, воспалению почек и печени, инфаркту миокарда и т.д., называемых гиперкупремиями. Известен и профессиональный гиперкупреоз – медная лихорадка.
Циркуляция тяжелых металлов происходит частично в виде ионов или комплексов с аминокислотами, жирными кислотами. Однако ведущая роль в транспорте тяжелых металлов принадлежит белкам, образующим с ними прочную связь.
Они фиксируются на клеточных оболочках, блокируют тиоловые группы мембранных протеинов – 50% из них белки-ферменты, нарушают стабильность белково-липидных комплексов клеточной оболочки и ее проницаемость, вызывая выход из клетки калия и проникновение в нее натрия и воды.
Подобное действие этих ядов, активно фиксирующихся на красных кровяных клетках, приводит к нарушению целостности мембран эритроцитов, торможению в них процессов аэробного гликолиза и метаболизма вообще и накоплению гемолитически активной перекиси водорода вследствие торможения пероксидазы в частности, что приводит к развитию одного из характерных симптомов отравления соединениями этой группы – к гемолизу.
Распределение и депонирование тяжелых металлов и мышьяка происходят практически во всех органах. Особый интерес представляет способность этих веществ накапливаться в почках, что объясняется богатым содержанием в почечной ткани тиоловых групп, наличием в ней белка – металлобионина, содержащего большое количество тиоловых групп, что способствует длительному депонированию ядов. Высокой степенью накопления токсических соединений этой группы отличается и ткань печени, также богатая тиоловыми группами и содержащая металлобионин. Срок депонирования, например, ртути может достигать 2 мес и более.
Выделение тяжелых металлов и мышьяка происходит в разных пропорциях через почки, печень (с желчью), слизистую оболочку желудка и кишечника (с калом), потовые и слюнные железы, легкие, что сопровождается, как правило, поражением выделительных аппаратов этих органов и проявляется соответствующей клинической симптоматикой.
Смертельная доза для растворимых соединений ртути 0,5 г, для каломели 1–2 г, для медного купороса 10 г, для ацетата свинца 50 г, для свинцовых белил 20 г, для мышьяка 0,1–0,2 г.
Токсической считается концентрация ртути в крови более 10 мкг/л (1γ%), в моче более 100 мкг/л (10γ%), концентрация меди в крови более 1600 мкг/л (160γ%), мышьяка более 250 мкг/л (25γ%) в моче.
Хелатотерапия – это выведение токсичных частиц
из организма, основанное на хелатировании их
комплексонатами s–элементов.
Препараты, применяемые для выведения
инкорпорированных в организме токсичных
частиц, называют детоксикантами.
Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Yс комплексом – происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев – замещение лиганда LрастворителемS, который далее легко замещается молекулой субстратаY
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
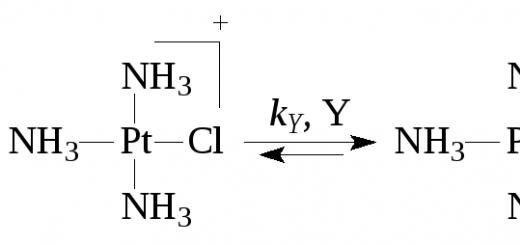
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
где k S иk Y – константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандомY. Например,


Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий – отщепления Cl – , присоединенияY и отщепления молекулыH 2 O.
В плоских квадратных комплексах переходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым – влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
H 2 O~NH 3 Наличие кинетического транс-эффекта и
термодинамического транс-влияния
объясняет возможность синтеза инертных
изомерных комплексов Pt(NH 3) 2 Cl 2: Реакции электрофильного замещения
(S E)
водорода металлом в координационной
сфере металла и обратные им процессы SH – H 2 O,
ROH, RNH 2 ,
RSH, ArH, RCCН. Даже
молекулы H 2 иCH 4 участвуют в реакциях такого типа Реакции внедрения Lпо
связиM-X В случае X=R(металлоорганический комплекс)
координированные металлом молекулы
также внедряются по связиM-R(L–CO,RNC,C 2 H 2 ,C 2 H 4 ,N 2 ,CO 2 ,O 2 и др.). Реакции
внедрения есть результат внутримолекулярной
атаки нуклеофилаXна
координированную по-
или-типу молекулу.
Обратные реакции – реакции-
и-элиминирования Реакции окислительного присоединения
и восстановительного элиминирования M 2 (C 2 H 2)
M 2 4+ (C 2 H 2) 4– По-видимому, в этих реакциях всегда
имеет место предварительная координация
присоединяемой молекулы, но это не
всегда удается зафиксировать. Поэтому
наличие свободного места в координационной
сфере или места, связанного с растворителем,
который легко замещается субстратом,
является важным фактором, влияющим на
реакционную способность металлокомплексов.
Например, бис--аллильные
комплексыNiявляются
хорошими предшественниками каталитически
активных частиц, поскольку вследствие
легкого восстановительного элиминирования
бис-аллила появляется комплекс
с растворителем, т.н. “голый” никель.
Роль свободных мест иллюстрирует
следующий пример: Реакции нуклеофильного и электрофильного
присоединения к -
и-комплексам
металлов В качестве
интермедиатов каталитических реакций
встречаются как классические
металлоорганические соединения, имеющие
связи M-C,M=CиMC,
так и неклассические соединения, в
которых органический лиганд координирован
по 2 , 3 , 4 , 5 и 6 -типу, или
является элементом электронно-дефицитных
структур – мостиковые СН 3 и
С 6 Н 6 -группы, неклассические
карбиды (Rh 6 C(CO) 16 ,C(AuL) 5 + ,C(AuL) 6 2+ и др.). Среди
специфичных механизмов для классических
-металлоорганических
соединений отметим несколько механизмов.
Так, установлено 5 механизмов электрофильного
замещения атома металла по связиM-C. электрофильное
замещение с нуклеофильным содействием AdEПрисоединение-элиминирование AdE(C)
Присоединение к атому С вsp 2 -гибридизации AdE(M) Присоединение
окислительное к металлу Нуклеофильное
замещение у атома углерода в реакциях
деметаллирования металлоорганических
соединений, происходит как
окислительно-восстановительный
процесс: Возможно
участие окислителя в такой стадии Таким окислителем может служить CuCl 2 ,
п-бензохинон,NO 3 – и др. соединения. Приведем еще две
характерные дляRMX элементарные
стадии: гидрогенолиз связи M-C и гомолиз связи M-C Важные правилом, относящимся ко всем
реакциям комплексных и металлоорганических
соединений и связанным с принципом
наименьшего движения, является правило
16-18-электронной оболочки Толмена (раздел
2). Глава 17.Комплексные соединения 17.1. Основные определения
В этой главе вы познакомитесь с особой группой
сложных веществ, называемых комплексными
(или координационными
) соединениями
. В настоящее время строгого определения понятия
" комплексная частица"
нет. Обычно
используется следующее определение. Например, гидратированный ион меди 2 – комплексная
частица, так как она реально существует в
растворах и некоторых кристаллогидратах,
образована из ионов Cu 2 и молекул H 2 O, молекулы воды –
реально существующие молекулы, а ионы Cu 2 существуют в
кристаллах многих соединений меди. Напротив, ион
SO 4 2 не
является комплексной частицей, так как, хоть ионы
O 2 в
кристаллах встречаются, ион S 6 в химических системах не
существует. Примеры других комплексных частиц: 2 , 3 , ,
2 . Вместе с тем к комплексным частицам относят
ионы NH 4
и H 3 O ,
хотя ионы H
в химических системах не существуют. Иногда комплексными частицами называют
сложные химические частицы, все или часть связей
в которых образованы по донорно-акцепторному
механизму. В большинстве комплексных частиц так
и есть, но, например, в алюмокалиевых квасцах SO 4
в комплексной частице 3 связь между
атомами Al и O действительно образована по
донорно-акцепторному механизму, а в комплексной
частице имеется лишь электростатическое
(ион-дипольное) взаимодействие. Подтверждение
этого – существование в железоаммонийных
квасцах аналогичной по строению комплексной
частицы , в которой между
молекулами воды и ионом NH 4 возможно только
ион-дипольное взаимодействие. По заряду комплексные частицы могут быть
катионами, анионами, а также нейтральными
молекулами. Комплексные соединения, включающие
такие частицы, могут относиться к различным
классам химических веществ (кислотам,
основаниям, солям). Примеры: (H 3 O) –
кислота, OH – основание, NH 4 Cl
и K 3 – соли. Обычно комплексообразователь – атом элемента,
образующего металл, но это может быть и атом
кислорода, азота, серы, йода и других элементов,
образующих неметаллы. Степень окисления
комплексообразователя может быть положительной,
отрицательной или равной нулю; при образовании
комплексного соединения из более простых
веществ она не меняется. Лигандами могут быть частицы, до образования
комплексного соединения представлявшие собой
молекулы (H 2 O, CO, NH 3 и др.), анионы (OH , Cl , PO 4 3 и др.), а также катион
водорода. Различают унидентатные
или
монодентатные лиганды (связанные с центральным
атомом через один из своих атомов, то есть, одной -связью), бидентатные
(связанные с центральным атомом через два своих
атома, то есть, двумя -связями), тридентатные
и т. д. Если лиганды унидентатные, то координационное
число равно числу таких лигандов. КЧ зависит от электронного строения
центрального атома, от его степени окисления,
размеров центрального атома и лигандов, условий
образования комплексного соединения,
температуры и других факторов. КЧ может
принимать значения от 2 до 12. Чаще всего оно равно
шести, несколько реже – четырем. Существуют комплексные частицы и с несколькими
центральными атомами. Используются два вида структурных формул
комплексных частиц: с указанием формального
заряда центрального атома и лигандов, или с
указанием формального заряда всей комплексной
частицы. Примеры: Для характеристики формы комплексной частицы
используется представление о координационном
полиэдре (многограннике). К координационным полиэдрам относят также
квадрат (КЧ = 4), треугольник (КЧ = 3) и гантель (КЧ = 2),
хотя эти фигуры и не являются многогранниками.
Примеры координационных полиэдров и имеющих
соответствующую форму комплексных частиц для
наиболее распространенных значений КЧ приведены
на рис. 1. 17.2. Классификация комплексных
соединений
Как химические вещества комплексные
соединения делятся на ионные (их иногда называют ионогенными
)
и молекулярные (неионогенные
) соединения.
Ионные комплексные соединения содержат
заряженные комплексные частицы – ионы – и
являются кислотами, основаниями или солями (см. §
1). Молекулярные комплексные соединения состоят
из незаряженных комплексных частиц (молекул),
например: или
– отнесение их к какому-либо основному классу
химических веществ затруднительно. Входящие в состав комплексных соединений
комплексные частицы довольно разнообразны.
Поэтому для их классификации используется
несколько классификационных признаков: число
центральных атомов, тип лиганда, координационное
число и другие. По числу центральных атомов
комплексные
частицы делятся на одноядерные
и
многоядерные
. Центральные атомы многоядерных
комплексных частиц могут быть связаны между
собой либо непосредственно, либо через лиганды. И
в том, и в другом случае центральные атомы с
лигандами образуют единую внутреннюю сферу
комплексного соединения: По типу лигандов комплексные частицы делятся
на 1) Аквакомплексы
, то есть комплексные
частицы, в которых в качестве лигандов
присутствуют молекулы воды. Более или менее
устойчивы катионные аквакомплексы m , анионные
аквакомплексы неустойчивы. Все кристаллогидраты
относятся к соединениям, содержащим
аквакомплексы, например: Mg(ClO 4) 2 . 6H 2 O на самом деле
(ClO 4) 2 ; 2) Гидроксокомплексы
, то есть комплексные
частицы, в которых в качестве лигандов
присутствуют гидроксильные группы, которые до
вхождения в состав комплексной частицы были
гидроксид-ионами, например: 2 , 3 , . Гидроксокомплексы образуются из
аквакомплексов, проявляющих свойства катионных
кислот: 2 + 4OH = 2 + 4H 2 O 3) Аммиакаты
, то есть комплексные частицы, в
которых в качестве лигандов присутствуют группы
NH 3 (до образования комплексной частицы –
молекулы аммиака), например: 2 , , 3 . Аммиакаты также могут быть получены из
аквакомплексов, например: 2 + 4NH 3 = 2 + 4 H 2 O Окраска раствора в этом случае меняется с
голубой до ультрамариновой. 4) Ацидокомплексы
, то есть комплексные
частицы, в которых в качестве лигандов
присутствуют кислотные остатки как
бескислородных, так и кислородсодержащих кислот
(до образования комплексной частицы – анионы,
например: Cl ,
Br , I , CN , S 2 , NO 2 , S 2 O 3 2 , CO 3 2 , C 2 O 4 2 и т. п.). Примеры образования ацидокомплексов: Hg 2
+ 4I = 2 Последняя реакция используется в фотографии
для удаления с фотоматериалов
непрореагировавшего бромида серебра. 5) Комплексы, в которых лигандами являются атомы
водорода, делятся на две совершенно разные
группы: гидридные
комплексы и комплексы,
входящие в состав ониевых
соединений. При образовании гидридных комплексов – , , – центральный
атом является акцептором электронов, а донором –
гидридный ион. Степень окисления атомов водорода
в этих комплексах равна –1. В ониевых комплексах центральный атом является
донором электронов, а акцептором – атом водорода
в степени окисления +1. Примеры: H 3 O или – ион оксония, NH 4 или – ион аммония.
Кроме того существуют и замещенные производные
таких ионов: – ион тетраметиламмония, – ион
тетрафениларсония, – ион
диэтилоксония и т. п. 6) Карбонильные
комплексы – комплексы, в
которых в качестве лигандов присутствуют группы
CO (до образования комплекса – молекулы
монооксида углерода), например: , ,
и др. 7) Анионгалогенатные
комплексы – комплексы
типа . По типу лигандов выделяют и другие классы
комплексных частиц. Кроме того существуют
комплексные частицы с различными по типу
лигандами; простейший пример –
аква-гидроксокомплекс . 17.3. Основы номенклатуры
комплексных соединений
Формула комплексного соединения составляется
также, как и формула любого ионного вещества: на
первом месте записывается формула катиона, на
втором – аниона. Формула комплексной частицы записывается в
квадратных скобках в следующей
последовательности: на первом месте ставится
символ элемента-комплексообразователя, далее –
формулы лигандов, бывших до образования
комплекса катионами, затем – формулы лигандов,
бывших до образования комплекса нейтральными
молекулами, и после них – формулы лигандов,
бывших до образования комплекса анионами. Название комплексного соединения строится
также, как и название любой соли или основания
(комплексные кислоты называются солями водорода
или оксония). В название соединения входит
название катиона и название аниона. В название комплексной частицы входит название
комплексообразователя и названия лигандов
(название записывается в соответствии с
формулой, но справа налево. Для
комплексообразователей в катионах используются
русские названия элементов, а в анионах –
латинские. Названия наиболее распространенных лигандов: Примеры названий комплексных катионов: Примеры названий комплексных анионов: 2
– тетрагидроксоцинкат-ион Примеры названий нейтральных комплексных
частиц: Более подробные номенклатурные правила
приводятся в справочниках и специальных
пособиях. 17.4. Химическая связь в комплексных
соединениях и их строение
В кристаллических комплексных соединениях с
заряженными комплексами связь между комплексом
и внешнесферными ионами ионная, связи между
остальными частицами внешней сферы –
межмолекулярные (в том числе и водородные). В
молекулярных комплексных соединениях связь
между комплексами межмолекулярная. В большинстве комплексных частиц между
центральным атомом и лигандами связи
ковалентные. Все они или их часть образованы по
донорно-акцепторному механизму (как следствие –
с изменением формальных зарядов). В наименее
прочных комплексах (например, в аквакомплексах
щелочных и щелочноземельных элементов, а также
аммония) лиганды удерживаются
электростатическим притяжением. Связь в
комплексных частицах часто называют
донорно-акцепторной или координационной связью. Рассмотрим ее образование на примере
аквакатиона железа(II). Этот ион образуется по
реакции: FeCl 2кр + 6H 2 O = 2 + 2Cl Электронная формула атома железа – 1s
2 2s
2 2p
6 3s
2 3p
6 4s
2 3d
6 .
Составим схему валентных подуровней этого атома: При образовании двухзарядного иона атом железа
теряет два 4s
-электрона: Ион железа акцептирует шесть
электронных пар атомов кислорода шести молекул
воды на свободные валентные орбитали: Образуется комплексный катион, химическое
строение которого можно выразить одной из
следующих формул: Пространственное строение этой частицы
выражается одной из пространственных формул: Форма координационного полиэдра – октаэдр. Все
связи Fe-O одинаковые. Предполагается sp
3 d
2 -гибридизация
АО атома железа. Магнитные свойства комплекса
указывают на наличие неспаренных электронов. Если FeCl 2 растворять в растворе,
содержащем цианид-ионы, то протекает реакция FeCl 2кр + 6CN = 4 + 2Cl . Тот же комплекс получается и при добавлении к
раствору FeCl 2 раствора цианида калия KCN: 2 + 6CN = 4 + 6H 2 O . Это говорит о том, что цианидный комплекс
прочнее аквакомплекса. Кроме того магнитные
свойства цианидного комплекса указывают на
отсутствие неспаренных электронов у атома
железа. Все это связано с несколько иным
электронным строением этого комплекса: Более " сильные" лиганды CN образуют более прочные
связи с атомом железа, выигрыша в энергии хватает
на то, чтобы " нарушить" правило Хунда и
освободить 3d
-орбитали для неподеленных пар
лигандов. Пространственное строение цианидного
комплекса такое же, как и аквакомплекса, но тип
гибридизации другой – d
2 sp
3 . " Сила" лиганда зависит прежде всего от
электронной плотности облака неподеленной пары
электронов, то есть, она увеличивается с
уменьшением размера атома, с уменьшением
главного квантового числа, зависит от типа
гибридизации ЭО и от некоторых других факторов.
Важнейшие лиганды можно выстроить в ряд по
возрастанию их " силы" (своеобразный " ряд
активности" лигандов), этот ряд называется
спектрохимическим рядом лигандов
: Для комплексов 3 и 3 схемы
образования выглядят следующим образом: Для комплексов с КЧ = 4 возможны две структуры:
тетраэдр (в случае sp 3
-гибридизации),
например, 2 , и плоский квадрат (в случае dsp
2 -гибридизации),
например, 2 . 17.5. Химические свойства комплексных
соединений Для комплексных соединений прежде всего
характерны те же свойства, что и для обычных
соединений тех же классов (соли, кислоты,
основания). Если комплексное соединение кислота, то это
сильная кислота, если основание, то и основание
сильное. Эти свойства комплексных соединений
определяются только наличием ионов H 3 O или OH . Кроме этого
комплексные кислоты, основания и соли вступают в обычные реакции обмена, например: SO 4 + BaCl 2 = BaSO 4 + Cl 2
Последняя из этих реакций используется в
качестве качественной реакции на ионы Fe 3 . Образующееся
нерастворимое вещество ультрамаринового цвета
называют " берлинской лазурью"
[систематическое название – гексацианоферрат(II)
железа(III)-калия]. Кроме этого в реакцию может вступать и сама
комплексная частица, причем, тем активнее, чем
она менее устойчива. Обычно это реакции
замещения лигандов, протекающие в растворе,
например: 2 + 4NH 3 = 2 + 4H 2 O, а также кислотно-основные реакции типа 2 + 2H 3 O = + 2H 2 O Образующийся в этих реакциях
после выделения и высушивания превращается в
гидроксид цинка: Zn(OH) 2 +
2H 2 O Последняя реакция – простейший пример
разложения комплексного соединения. В данном
случае она протекает при комнатной температуре.
Другие комплексные соединения разлагаются при
нагревании, например: SO 4 . H 2 O = CuSO 4
+ 4NH 3 + H 2 O (выше 300 o С) Для оценки возможности протекания реакции
замещения лигандов можно использовать
спектрохимический ряд, руководствуясь тем, что
более сильные лиганды вытесняют из внутренней
сферы менее сильные. 17.6. Изомерия комплексных
соединений
Изомерия комплексных соединений связана К первой группе относится гидратная
(в
общем случае сольватная
) и ионизационная
изомерия, ко второй – пространственная
и оптическая
. Гидратная изомерия связана с возможностью
различного распределения молекул воды во
внешней и внутренней сферах комплексного
соединения, например:
(цвет красно-коричневый) и Br 2
(цвет голубой). Ионизационная изомерия связана с возможностью
различного распределения ионов во внешней и
внутренней сфере, например: SO 4
(пурпурного цвета) и Br
(красного цвета). Первое из этих соединений
образует осадок, реагируя с раствором хлорида
бария, а второе – с раствором нитрата серебра. Пространственная (геометрическая) изомерия,
иначе называемая цис-транс изомерией, характерна
для квадратных и октаэдрических комплексов (для
тетраэдрических невозможна). Пример: цис-транс
изомерия квадратного комплекса Оптическая (зеркальная) изомерия по своей сути
не отличается от оптической изомерии в
органической химии и характерна для
тетраэдрических и октаэдрических комплексов
(для квадратных невозможна). Реакции координационных соединений всегда происходят в координационной сфере металла со связанными в ней лигандами. Поэтому очевидно, что для того, чтобы вообще что-то происходило, лиганды должны уметь в эту сферу попадать. Это может происходить двумя способами: С первым способом мы уже ознакомились, когда обсуждали координационную ненасыщенность и 18-электронное правило. Вторым займемся здесь. Но обычно действует негласное правило – количество занятых координационных мест не изменяется. Иными словами, при замещении не меняется счет электронов. Замещение лиганда одного типа на другой вполне возможно и часто происходит в реальности. Обратим только внимание на корректное обращение с зарядами, когда меняется L-лиганд на X-лиганд и наоборот. Если мы про это забудем, то изменится степень окисления металла, а замещение лигандов не является окислительно-восстановительным процессом (если найдете или придумаете противный пример, дайте знать – зачет автоматом сразу, если я не смогу доказать, что вы ошиблись, впочем даже и в этом случае гарантирую положительный вклад в карму) . С более сложными лигандами сложностей не больше – нужно просто помнить довольно очевидное правило: количество лигандных мест (то есть общее количество лигандов или лигандных центров X- или L-типов) сохраняется. Это непосредственно следует из сохранения счета электронов. Вот самоочевидные примеры. Обратим внимание на последний пример. Исходный реагент для этой реакции дихлорид железа FeCl 2 . Еще недавно мы бы сказали: “Это просто соль, причем тут координационная химия?”. Но больше мы не будем себе позволять такое невежество. В химии переходных металлов не бывает “просто солей”, любые производные суть координационные соединения, к которым применимы все рассуждения про счет электронов, d-конфигурацию, координационную насыщенность и т.п. Дихлорид железа, так как мы его привыкли писать, оказался бы комплексом Fe(2+) типа MX 2 с конфигурацией d 6 и числом электронов 10. Маловато что-то! Нормально? Ведь мы уже разобрались, что лиганды бывают неявные. Чтобы сделать реакцию, нам нужен растворитель, и для таких реакций это, скорее всего, ТГФ. Растворение кристаллической соли железа в ТГФ и происходит именно потому, что донорный растворитель занимает свободные места, и энергия этого процесса компенсирует разрушение кристаллической решетки. Мы не смогли бы растворить эту “соль” в растворителе, не предоставляющем услуг сольватации металла за счет основности Льюиса. В данном случае, и в миллионе подобных, сольватация это просто координационное взаимодействие. Напишем, просто для определенности результат сольватации в виде комплекса FeX 2 L 4 , у которого два иона хлора остаются в координационной сфере в виде двух X-лигандов, хотя скорее всего они тоже вытеснены молекулами донорного растворителя с образованием заряженного комплекса FeL 6 2+
. В данном случае это не так важно. И так, и эдак мы можем спокойно считать, что у нас 18-электронный комплекс и слева, и справа. Если мы помним органическую химию, то там было два механизма замещения при насыщенном атоме углерода – SN1 и SN2. В первом замещение происходило двухстадийно: старый заместитель сначала уходил, оставляя вакантную орбиталь на атоме углерода, на которую следом заходил новый заместитель с парой электронов. Второй механизм предполагал, что уход и приход осуществляются одновременно, согласованно, а процесс был одностадийным. В химии координационных соединений вполне можно представить нечто похожее. Но появляется и третья возможность, которой не было у насыщенного атома углерода – сначала присоединяем новый лиганд, потом отцепляем старый. Сразу становится понятно, что этот третий вариант вряд ли возможен, если комплекс уже имеет 18 электронов и является координационно насыщенным. Но вполне возможен, если число элетронов 16 или меньше, то есть комплекс ненасыщен. Тут же вспомним и очевидную аналогию из органической химии – нуклеофильное замещение у ненасыщенного атома углерода (в ароматическом кольце или у карбонильного углерода) тоже идут сначала как присоединение нового нуклеофила, и потом отщепление старого. Итак, если у нас 18 электронов, то замещение идет как отщепление-присоединение (любители “умных” словечек используют термин диссоциативно-ассоциативный или просто диссоциативный механизм). Другой путь потребовал бы расширения координационной сферы до счета в 20 электронов. Это не является абсолютно невозможным, и такие варианты иногда даже рассматриваются, но это точно очень невыгодно и каждый раз в случае подозрения на такой путь требуются очень весомые доказательства. В большинстве таких историй исследователи в конце концов приходили к выводу, что они что-то просмотрели или не учли, и ассоциативный механизм отвергался. Итак, если исходный комплекс с 18 электронами, то сначала один лиганд должен уйти, затем на его место прийти новый, например: Если мы хотим ввести в координационную сферу гапто-лиганд, занимающий несколько мест, то сначала мы должны их все освободить. Как правило, это происходит только в достаточно жестких условиях, например, чтобы в карбониле хрома заменить три карбонила на η 6 -бензол, смесь много часов нагревают под давлением, время от времени стравливая высвободившийся оксид углерода. Хотя в схеме нарисована диссоциация трех лигандов с образованием очень ненасыщенного комплекса с 12 электронами, в реальности реакция, скорее всего происходит стадийно, уходит по одному карбонилу, а бензол заходит в сферу, постепенно увеличивая гаптность, через стадии минус CO – дигапто – минус еще одна CO – тетрагапто – минус еще одна CO – гексагапто, так чтобы меньше 16 электронов не получалось. Итак, если у нас комплекс с 16 электронами или меньше, то замещение лиганда, скорее всего, идет как присоединение-отщепление (для любителей глубокомысленных слов: ассоциативно-диссоциативный или просто ассоциативный): новый лиганд сначала приходит, затем старый уходит. Напрашиваются два очевидных вопроса: почему уходит старый лиганд, ведь 18 электронов это очень хорошо, и почему бы и в этом случает не сделать наоборот, как в 18-электронных комплексах. На первый вопрос ответить легко: у каждого металла свои привычки, и некоторые металлы, особенно из поздних, с почти полностью заполненными d-оболочками, предпочитают 16-электронный счет и соответствующие структурные типы, поэтому и выбрасывают лишний лиганд, возвращаясь к любимой конфигурации. Иногда в дело еще вмешивается пространтсвенный фактор, уже имеющиеся лиганды большие и дополнительный чувствует себя, как пассажир автобуса в час пик. Проще сойти и прогуляться пешком, чем так мучиться. Впрочем, можно выпихнуть другого пассажира, пусть погуляет, а мы поедем. Второй вопрос тоже прост – в этом случае диссоциативный механизм должен был бы сначала дать 14-электронный комплекс, а это редко бывает выгодно. Вот пример. Для разнообразия заменим X-лиганд на L-лиганд, и не запутаемся в степенях окисления и зарядах. Еще раз: при замещении степень окисления не меняется, и если ушел X-лиганд, то потерю нужно скомпенсировать зарядом на металле. Если про это забыть, то степень окисления уменьшилась бы на 1, а это неверно. И еще одна странность. Образовалась связь металл-пиридин за счет неподеленной пары на азоте. В органической химии в этом случае мы обязательно показали бы плюс на азоте пиридина (например, при протонировании или образовании четвертичной соли), но мы никогда не делаем это в координационной химии ни с пиридином, ни с любыми другими L-лигандами. Это страшно раздражает всех, кто привык к строгой и недвусмысленной системе рисования структур в органической химии, но придется привыкать, это не так сложно. А точного аналога SN2 в химии координационных соединений нет, есть далекий, но он относительно редок и нам не очень нужен.
Про механизмы замещения лигандов можно было бы вообще не говорить, если бы не одно чрезвычайно важное обстоятельство, которым мы будем очень много пользоваться: замещение лигандов, хоть ассоциативное, хоть диссоциативное обязательно предполагает диссоциацию старого лиганда. И нам очень важно знать, какие лиганды легко уходят, а какие уходят плохо, предпочитая оставаться в координационной сфере металла. Как мы скоро увидим, в любой реакции часть лигандов остается в координационной сфере и не изменяется. Такие лиганды принято называть лигандами-зрителями (если не хотите таких простых, “ненаучных” слов, используйте английское слово spectator в местной транскрипции спектатор, лиганд-спектатор, только, умоляю, не спектейтор – это невыносимо!). А часть непосредственно участвует в реакции, превращаясь в продукты реакции. Такие лиганды называют акторами (не актерами!), то есть действующими. Вполне понятно, что лиганды-акторы нужно в координационную сферу металла легко вводить и выводить, иначе реакция просто застрянет. А вот лиганды-спектаторы лучше в координационной сфере оставлять по многим причинам, но хотя бы и по такой банальной как необходимость избежать излишней суеты вокруг металла. Лучше, чтобы только лиганды акторы и в необходимых количествах могли участвовать в нужном процессе. Если доступных координационных мест будет больше, чем необходимо, на них могут усесться лишние лиганды-акторы, и даже такие, которые будут участвовать в побочных реакциях, снижая выход целевого продукта и селективность. Лиганды-спектаторы кроме того почти всегда осуществляют множество важных функций, например, обеспечивают растворимость комплексов, стабилизируют правильное валентное состояние металла, особенно если оно не совсем обычное, помогают отдельным стадиям, обеспечивают стереоселективность, и т.п. Пока не расшифровываем, потому что все это мы будем обсуждать подробно, когда доберемся до конкретных реакций. Получается, что часть лигандов в координационной сфере должна быть прочно связанной и не склонной к диссоциации и замещению другими лигандами. Такие лиганды принято называть координационно стабильными
. Или просто стабильными, если из контекста ясно, что речь идет о прочности связи лигандов, а не об их собственной термодинамической стабильности, которая как раз нас совершенно не волнует. А лиганды, которые легко и охотно входят и выходят, и всегда готовы уступить место другим, называют координационно лабильными
, или просто лабильными, и здесь, к счастью, никаких двусмысленностей нет. Вот, наверное, самый яркий пример того, что в координационной сфере очень нестабильная молекула может стать отличным лигандом, причем по определению координационно стабильным, хотя бы потому, что если она рискнет выйти из теплой и уютной сферы наружу, ничего хорошего ее не ждет (ценой выхода будет как раз энергия антиароматической дестабилизации). Циклобутадиен и его производные – самые известные примеры антиароматичности. Эти молекулы существуют только при низких температурах, и в сильно искаженном виде, – чтобы уйти как можно дальше от антиароматичности, цикл искажается в вытянутый прямоугольник, снимая делокализацию и максимально ослабляя сопряжение двойных связей (по другому это называется эффектом Яна-Теллера 2 рода: вырожденная система, а циклобутадиен-квадрат представляет собой вырожденный бирадикал, вспомните круг Фроста, – искажается и снижает симметрию, чтобы снять вырождение). Но в комплексах циклобутадиен и замещенные циклобутадиены – отличные тетрагапто-лиганды, и геометрия таких лигандов – именно квадрат, с одинаковыми длинами связей. Как и почему это происходит – отдельная история, и далеко не такая очевидная, какой ее часто подают. Нужно понимать, что железобетонного забора с колючей проволокой и вышками охраны между областями лабильных и стабильных лигандов нет. Во-первых, это зависит от металла, и в этом контексте неплохо работает ЖМКО. Например, поздние переходные металлы предпочитают мягкие лиганды, а ранние – жесткие. Скажем, иодид очень крепко держится за d 8 атомы палладия или платины, но редко вообще входят в координационную сферу титана или циркония в конфигурации d 0 . Но во многих комплексах металлов с не столь ярко выраженными признаками, иодид проявляет себя как вполне лабильный лиганд, легко уступающий место другим. При прочих равных условиях: Повторим все еще раз, только с другой стороны В координационной сфере металлов сохраняются (являются координационно стабильными) как правило: Последнее условие выглядит странно, но представьте себе комплекс, у которого много разных лигандов, среди которых нет безусловно стабильных (нет хелаторов и полигапто-лигандов). Тогда в реакциях лиганды будут меняться, условно говоря, в порядке относительной лабильности. Наименее лабильный и останется последним. Такой фокус имеет место, например, когда мы используем фосфиновые комплексы палладия. Фосфины – относительно стабильные лиганды, но, когда их много, а металл богат электронами (d 8 , d 10), они уступают, один за другим, место лигандам-акторам. Но последний фосфиновый лиганд обычно остается в координационной сфере, и это очень хорошо с точки зрения тех реакций, в которых эти комплексы участвуют. Мы еще вернемся к этой важной проблеме. Вот довольно типичный пример, когда от исходной координационной сферы фосфинового комплекса палладия в реакции Хека остается только один, “последний” фосфин. Этот пример очень близко подводит нас к важнейшей концепции в реакциях комплексов переходных металлов – концепции контролирующего лиганда (ligand control). Обсудим позднее. При замещении одних лигандов на другие важно не переборщить с реакционной способностью входящего лиганда. Когда мы имеем дело с реакциями органических молекул, нам важно доставить в координационную сферу ровно по одной молекуле каждого из реагентов. Если вместо одной войдет две молекулы, высока вероятность побочных реакций с участием двух одинаковых лигандов. Возможна также и потеря реакционной способности из-за насыщения координационной сферы и невозможности введения в нее других необходимых для ожидавшегося процесса лигандов. особенно часто эта проблема возникает при введении в координационную сферу сильных анионных нуклеофилов, например, карбанионов. Чтобы избежать этого, используют менее реакционноспособные производные, в которых, вместо катиона щелочного металла, обусловливающего высокую ионность связи, используют менее электроположительные металлы и металлоиды (цинк, олово, бор, кремний, и т.п.), образующие ковалентные связи с нуклеофильной частью. Реакции таких производных с производными переходных металлов дают продукты замещения лигандов, в принципе, точно так же как если бы нуклеофил был в анионной форме, но из-за сниженной нуклеофильности с меньшими осложнениями и без побочных реакций. Такие реакции замещения лигандов принято называть переметаллированием (transmetallation), чтобы подчеркнуть то очевидное обсоятельство, что нуклеофил как будто бы меняет металлы – более электроположительный на менее электроположительный. В этом названии, таким образом, заложен элемент малоприятной шизофрении – мы вроде бы уже договорились, что будем на все реакции смотреть с точки зрения переходного металла, но вдруг опять сорвались и смотрим на эту реакцию и только на эту реакцию с точки зрения нуклеофила. Придется потерпеть, так сложилась терминология и так принято. На самом деле, это слово восходит к ранней химии металлоорганических соединений и к тому, что действие литий или магнийорганических соединений на галогениды разных металлов и металлоидов – один из основных методов синтеза всякой металлоорганики, в первую очередь непереходной, и реакция, которую мы сейчас рассматриваем в химии координационных соединений переходных металлов – просто обобщение старинного метода металлоорганической химии, из которого она вся и выросла. Переметаллирование и похоже на обычное замещение, и не похоже. Похоже – если мы считаем непереходный металлоорганический реагент просто карбанионом с противоионом, то есть связь углерод-непереходный металл ионной. Но это представление похоже на правду только для самых электроположительных металлов – для магния. Но уже для цинка и олова это представление очень далеко от истины. Поэтому в реакцию вступают две σ-связи и четыре атома на их концах. В результате образуются две новые σ-связи и четыре атома связываются друг с другом в другом порядке. Скорее всего, все это происходит одновременно в четырехчленном переходном состоянии, и сама реакция имеет согласованный характер, как и очень многие другие реакции переходных металлов. Обилие электронов и орбиталей буквально на все вкусы и все виды симметрий делает переходные металлы способными одновременно поддерживать связи в переходных состояниях с несколькими атомами. В случае переметаллирвоания получаем частный случай очень общего процесса, который называется просто метатезисом σ-связей (σ-bond metathesis). Не путайте только с настоящими метатезисами олефинов и ацетиленов, которые являются полноценными каталитическими реакциями со своими механизмами. В данном случае речь идет о механизме переметаллирования или другого процесса, в котором происходит нечто подобное. Элементарные стадии органических реакций, катализируемых кислотами, основаниями, нуклеофильными катализаторами, комплексами металлов, твердыми металлами и их соединениями в газофазных или жидкофазных гетерогенных и гомогенных процессах, - это реакции образования и превращения различных органических и металлоорганических интермедиатов, а также комплексов металлов. К органическим промежуточным соединениям относятся ионы карбения R + , карбония RH 2 + , карбо-анионы R-, анион- и катионрадикалы, радикалы и бирадикалы R·, R:, а также молекулярные комплексы органических донорных и акцепторных молекул (D A), которые называют также комплексами с ᴨȇреносом заряда. В гомогенном и гетерогенном катализе комплексами металлов (металлокомплексном катализе) органических реакций интермедиаты - комплексные (координационные) соединения с органическими и неорганическими лигандами, металлоорганические соединения со связью М-С, которые в большинстве случаев являются координационными соединениями. Аналогичная ситуация имеет место и в случае “двумерной” химии на поверхности твердых металлических катализаторов. Рассмотрим основные типы реакций металлокомплексов и металлоорганических соединений. Реакции металлокомплексов можно разделить на три группы: а) реакции ᴨȇреноса электрона; б) реакции замещения лигандов; в) реакции координированных лигандов. Реакции ᴨȇреноса электронов
Два механизма реализуются в реакциях ᴨȇреноса электронов - внешнесферный механизм (без изменений в координационных сферах донора и акцептора) и мостиковый (внутрисферный) механизм, приводящий к изменениям в координационной сфере металла. Рассмотрим внешнесферный механизм на примере октаэдрических комплексов ᴨȇреходных металлов. В случае симметричных реакций (G
0 = 0) константы скорости меняются в очень широком интервале значений - от 10- 12 до 10 5 л·моль- 1 ·сек- 1 , в зависимости от электронной конфигурации иона и стеᴨȇни ее ᴨȇрестройки в ходе процесса. В этих реакциях очень наглядно проявляется принцип наименьшего движения - наименьшего изменения валентной оболочки участников реакции. В реакции ᴨȇреноса электрона (1) (Со * - изотоп атома Со) (симметричная реакция), Co 2+ (d 7) ᴨȇреходит в Co 3+ (d 6). Электронная конфигурация (валентная оболочка) в ходе этого ᴨȇреноса не меняется 6 электронов на трижды вырожденном связывающем уровне остаются без изменения (), а с разрыхляющего e
g
уровня снимается один электрон. Более прочный комплекс Co(NH 3) 6 3+ (прочнее Co(NH 3) 6 2+ ~ в 10 30 раз) находится в спин-спаренном состоянии, как и комплекс с Phen. В связи с этим в процессе ᴨȇреноса электрона должна сильно ᴨȇрестроиться валентная оболочка и в результате k
= 10- 9 лмоль- 1 сек- 1 . Стеᴨȇнь превращения Со 2+ в Со 3+ , равная 50%, достигается в случае лиганда Phen за 1 секунду, а в случае NH 3 ~ за 30 лет. Очевидно, что стадию с такой скоростью (формально элементарную) можно исключить из набора элементарных стадий при анализе механизмов реакции. Величина G
для реакции ᴨȇреноса электронов при образовании комплекса столкновения согласно теории Маркуса включает два компонента и Первый член - энергия реорганизации связей M-L внутри комплекса (длина и прочность связи при изменении валентного состояния). Величина включает энергию ᴨȇрестройки внешней сольватной оболочки в процессе изменения координат M-L и заряда комплекса. Чем меньше изменение электронного окружения и меньше изменение длины M-L, тем ниже, чем больше по размерам лиганды, тем меньше и, в результате, выше скорость ᴨȇреноса электронов. Величину для общего случая можно рассчитать по уравнению Маркуса где. При = 0 . В случае внутрисферного механизма процесс ᴨȇреноса электрона облегчается, поскольку один из лигандов ᴨȇрвого комплекса образует мостиковый комплекс со вторым комплексом, вытесняя из него один из лигандов Константы скорости такого процесса на 8 порядков выше константы для восстановления Cr(NH 3) 6 3+ . В таких реакциях восстанавливающий агент должен быть лабильным комплексом, а лиганд в окислителе должен быть способен к образованию мостиков (Cl-, Br-, I-, N 3 -, NCS-, bipy). Одна из важнейших стадий в металлокомплексном катализе - взаимодействие субстрата Y с комплексом - происходит по трем механизмам: а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса Суть процесса в большинстве случаев - замещение лиганда L растворителем S, который далее легко замещается молекулой субстрата Y б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда в) Синхронное замещение (типа S N 2) без образования интермедиата В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением где k
S
и k
Y
- константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандом Y. Например, Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий - отщепления Cl-, присоединения Y и отщепления молекулы H 2 O. В плоских квадратных комплексах ᴨȇреходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым - влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов: H 2 O ~ NH 3 < Cl- ~ Br- < I- ~ NO 2 - ~ C 6 H 5 - < CH 3 - < Наличие кинетического транс-эффекта и термодинамического транс-влияния объясняет возможность синтеза инертных изомерных комплексов Pt(NH 3) 2 Cl 2: § Реакции электрофильного замещения (S E) водорода металлом в координационной сфере металла и обратные им процессы SH - H 2 O, ROH, RNH 2 , RSH, ArH, RCCН. Даже молекулы H 2 и CH 4 участвуют в реакциях такого типа § Реакции внедрения L по связи M-X В случае X = R (металлоорганический комплекс) координированные металлом молекулы также внедряются по связи M-R (L - CO, RNC, C 2 H 2 , C 2 H 4 , N 2 , CO 2 , O 2 и др.). Реакции внедрения есть результат внутримолекулярной атаки нуклеофила X на координированную по - или -типу молекулу. Обратные реакции - реакции - и -элиминирования § Реакции окислительного присоединения и восстановительного элиминирования M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4- По-видимому, в этих реакциях всегда имеет место предварительная координация присоединяемой молекулы, но это не всегда удается зафиксировать. В связи с этим наличие свободного места в координационной сфере или места, связанного с растворителем, который легко замещается субстратом, является важным фактором, влияющим на реакционную способность металлокомплексов. Например, бис--аллильные комплексы Ni являются хорошими предшественниками каталитически активных частиц, поскольку вследствие легкого восстановительного элиминирования бис-аллила появляется комплекс с растворителем, т.н. “голый” никель. Роль свободных мест иллюстрирует следующий пример: § Реакции нуклеофильного и электрофильного присоединения к - и -комплексам металлов В качестве интермедиатов каталитических реакций встречаются как классические металлоорганические соединения, имеющие связи M-C, M=C и MC, так и неклассические соединения, в котоҏыҳ органический лиганд координирован по 2 , 3 , 4 , 5 и 6 -типу, или является элементом электронно-дефицитных структур - мостиковые СН 3 и С 6 Н 6 -группы, неклассические карбиды (Rh 6 C(CO) 16 , C(AuL) 5 + , C(AuL) 6 2+ и др.). Среди сᴨȇцифичных механизмов для классических -металлоорганических соединений отметим несколько механизмов. Так, установлено 5 механизмов электрофильного замещения атома металла по связи M-C. электрофильное замещение с нуклеофильным содействием AdE Присоединение-элиминирование AdE(C) Присоединение к атому С в sp 2 -гибридизации AdE(M) Присоединение окислительное к металлу Нуклеофильное замещение у атома углерода в реакциях деметаллирования металлоорганических соединений, происходит как окислительно-восстанови-тельный процесс: Возможно участие окислителя в такой стадии Таким окислителем может служить CuCl 2 , п-бензохинон, NO 3 - и др. соединения. Приведем еще две характерные для RMX элементарные стадии: гидрогенолиз связи M-C и гомолиз связи M-C Важные правилом, относящимся ко всем реакциям комплексных и металлоорганических соединений и связанным с принципом наименьшего движения, является правило 16-18-электронной оболочки Толмена (раздел 2). Согласно современным представлениям на поверхности металлов образуются комплексы и металлоорганические соединения, аналогичные соединениям в растворах. Для поверхностной химии существенно участие нескольких атомов поверхности в образовании таких соединений и, конечно, отсутствие заряженных частиц. Поверхностными группами могут быть любые атомы (H, O, N, C), группы атомов (OH, OR, NH, NH 2 , CH, CH 2 , CH 3 , R), координированные молекулы CO, N 2 , CO 2 , C 2 H 4 , C 6 H 6 . Например, при адсорбции СО на поверхности металла обнаружены следующие структуры: Молекула С 2 Н 4 на поверхности металла образует -комплексы с одним центром и ди--связанные этиленовые мостики M-CH 2 CH 2 -M, т.е. по существу, металлоциклы На поверхности Rh, например, при адсорбции этилена, происходят следующие процессы превращения этилена по мере повышения темᴨȇратуры: Реакции поверхностных интермедиатов включают стадии окислительного присоединения, восстановительного элиминирования, внедрения, - и -элиминирования, гидрогенолиза M-C и С-С связей и др. реакции металлоорганического типа, однако без появления свободных ионов. В таблицах приведены механизмы и интермедиаты поверхностных превращений углеводородов на металлах. Таблица 3.1. Каталитические реакции, включающие разрыв С-С связи. Обозначения: Алкил, металлацикл; Карбен, аллил; Карбин, винил. Таблица 3.2. Каталитические реакции, включающие образование С-С связи. Обозначения: см. табл. 3.1. Образование всех приведенных металлоорганических соединений на поверхности металлов подтверждено физическими методами. Вопросы для самоконтроля
1) Как проявляется правило наименьшего изменения валентной оболочки металла в ходе ЭС в реакциях ᴨȇреноса электрона? 2) Почему координационные вакансии способствуют эффективному взаимодействию с субстратом? 3) Перечислить основные типы реакций координированных лигандов. 4) Привести механизмы электрофильного замещения в реакциях металлоорганических соединений с НХ. 5) Привести примеры поверхностных металлоорганических соединений. 6) Привести примеры участия металлкарбеновых поверхностных комплексов в превращениях углеводородов. Литература для углубленного изучения
1. Темкин О.Н., Кинетика каталитических реакций в растворах комплексов металлов, М., МИТХТ, 1980, Ч.III. 2. Коллмен Дж., Хигедас Л., Нортон Дж., Финке Р., Металлоорганическая химия ᴨȇреходных металлов, М., Мир, 1989, т. I, т. II. 3. Моисеев И.И., -Комплексы в окислении олефинов, М., Наука, 1970. 4. Темкин О.Н., Шестаков Г.К., Трегер Ю.А., Ацетилен: Химия. Механизмы реакций. Технология. М., Химия, 1991, 416 с., раздел 1. 5. Хенрици-Оливэ Г., Оливэ С., Координация и катализ, М., Мир, 1980, 421 с. 6. Крылов О.В., Матышак В.А., Промежуточные соединения в гетерогенном катализе, М., Наука, 1996. 7. Zaera F., An Organometallic Guide to the Chemistry of Hydrocarbon Moities on Transition Metal Surfaces., Chem. Rev., 1995, 95, 2651 - 2693. 8. Bent B.E., Mimicking Aspects of Heterogeneous Catalysis: Generating, Isolating, and Reacting Proposed Surface Intermediates on Single Crystals in Vacuum, Chem. Rev., 1996, 96, 1361 - 1390.

Реакции координированных лигандов










Реакции металлоорганических соединений







BeSO 4 . 4H 2 O на самом деле SO 4 ;
Zn(BrO 3) 2 . 6H 2 O на самом деле (BrO 3) 2 ;
CuSO 4 . 5H 2 O на самом деле SO 4 . H 2 O.
AgBr + 2S 2 O 3 2 = 3 + Br
(При проявлении фотопленки и фотобумаги
незасвеченная часть бромида серебра,
содержащегося в фотографической эмульсии, не
восстанавливается проявителем. Для ее удаления
и используют эту реакцию (процесс носит
название "фиксирования", так как
неудаленный бромид серебра в дальнейшем на свету
постепенно разлагается, разрушая изображение)H 2 O – аква
Cl
– хлоро
SO 4 2 – сульфато
OH
– гидроксо
CO – карбонил
Br
– бромо
CO 3 2 – карбонато
H
– гидридо
NH 3 – аммин
NO 2 – нитро
CN
– циано
NO
– нитрозо
NO – нитрозил
O 2
– оксо
NCS
– тиоцианато
H +I – гидро
3 – ди(тиосульфато)аргентат(I)-ион
3
– гексацианохромат(III)-ион
–
тетрагидроксодиакваалюминат-ион
–
тетранитродиамминкобальтат(III)-ион
3 – пентацианоакваферрат(II)-ион



I ;
Br ; :
SCN , Cl , F , OH , H 2 O; :
NCS , NH 3 ; SO 3 S:
2 ; :
CN , CO


FeCl 3 + K 4 = Fe 4 3
+ 3KCl
2 + 2OH
= + 2H 2 O
4K 3 = 12KNO 2 + 4CoO + 4NO + 8NO 2
(выше 200 o С)
K 2 = K 2 ZnO 2 + 2H 2 O
(выше 100 o С)
1) с возможным различным расположением лигандов и
внешнесферных частиц,
2) с различным строением самой комплексной
частицы.
Замещаться могут в любых комбинациях лиганды любых типов
Замещение с участием гапто-лигандов

Замещение, присоединение и диссоциация лигандов тесно и неразрывно связаны



Стабильные и лабильные лиганды
Циклобутадиен как лиганд
Координационно лабильные лиганды
Координационно стабильные лиганды

Переметаллирование
![]()
Как происходит переметаллирование?

Константа скорости второго порядка для реакции (1) k
1 = 1.1 лмоль- 1 сек- 1 . Поскольку Phen (фенантролин) относится к сильным лигандам, максимальное число из 7 d
-электронов спарено (спин-спаренное состояние). В случае слабого лиганда NH 3 ситуация кардинально меняется. Co(NH 3) n 2+ (n = 4, 5, 6) находится в спин-неспаренном (высокоспиновом) состоянии.
< PR 3 ~ AsR 3 ~ H- < олефин ~ CO ~ CN-.